Protein kinases function as pivotal regulators in biological events, governing essential cellular processes through the transfer of phosphate groups from ATP molecules to substrates. Dysregulation of kinase activity is frequently associated with cancer, ocasionally arising from chromosomal translocation events that relocate genes encoding kinases. Fusion proteins resulting from such events, particularly those involving the proto-oncogene tyrosine-protein kinase ROS (ROS1), manifest as constitutively active kinases, emphasizing their role in oncogenesis. Notably, the chromosomal reallocation of the ros1 gene leads to fusion of proteins with the ROS1 kinase domain, implicated in various cancer types. Despite their prevalence, targeted inhibition of these fusion proteins relies on repurposed kinase inhibitors. This review comprehensively surveys experimentally determined ROS1 structures, emphasizing the pivotal role of X-ray crystallography in providing high-quality insights. We delve into the intricate interactions between ROS1 and kinase inhibitors, shedding light on the structural basis for inhibition. Additionally, we explore point mutations identified in patients, employing molecular modeling to elucidate their structural impact on the ROS1 kinase domain. By integrating structural insights with in vitro and in silico data, this review advances our understanding of ROS1 kinase in cancer, offering potential avenues for targeted therapeutic strategies.
DOCUMENT
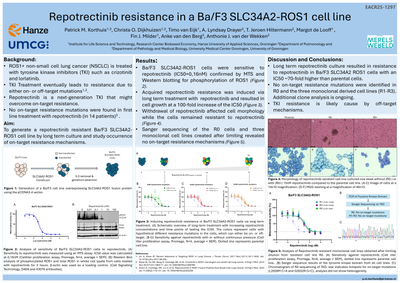
BackgroundThe ROS1 G2032R mutation is the most common on-target resistance mutation in crizotinib treated ROS1-positive lung cancer patients. The aim of our study was to investigate resistance mechanisms in SCL34A2-ROS1G2032R positive Ba/F3 cells against second line treatment with lorlatinib.MethodsBa/F3 SLC34A2-ROS1G2032R cells were subjected to N-ethyl-N-nitrosourea (ENU) mutagenesis and clones were selected upon treatment with 1000 nM lorlatinib for 4 weeks. Resistant clones were analyzed for presence of on-target resistant mutations using Sanger sequencing. In addition, we generated subclones expressing SLC34A2-ROS1L2026M+G2032R and SLC34A2-ROS1L2026M in Ba/F3 cells. Sensitivity to ROS1 TKIs was determined by measuring cell viability and ROS1 phosphorylation. Molecular Dynamic simulations of the ATP binding pocket were performed for all ROS1 variants.ResultsThe ENU-screen of 41 lorlatinib resistant clones revealed one with a mutation in the kinase domain: L2026M. Cell viability assays of the ENU-induced resistant cell line and the Ba/F3 cells transfected with the mutant SCL34A2-ROS1 fusion gene constructs revealed a decreased sensitivity of SLC34A2-ROS1L2026M+G2032R cells for lorlatinib, crizotinib, entrectinib and repotrectinib compared to the single mutants. Consistent with these findings, we observed phosphorylation of ROS1 fusion protein in the double mutant cells which was not inhibited upon treatment with ROS1 TKIs. The single mutant cells showed as expected a clear reduction in phosphorylated ROS1 fusion protein . Molecular modeling to unravel the effect of the mutations demonstrated that the volume of the ATP-binding pocket was reduced in single and double mutants compared to wild type. The double L2026M+G2032R mutant displayed the smallest pocket.ConclusionsWe identified a novel on-target mutation after inducing lorlatinib resistance in SLC34A2-ROS1G2032R Ba/F3 cells. This SLC34A2-ROS1L2026M+G2032R cell line was also resistant to crizotinib, entrectinib and repotrectinib. The resistance can be explained by a smaller ATP binding pocket in the mutated ROS1 fusion protein preventing effective binding of the investigated TKIs.
DOCUMENT
DOCUMENT

DOCUMENT

Clinical outcomes in ROS1-fusion positive (ROS1+) non-small cell lung cancer (NSCLC) by fusion partner and resistance mechanisms are limited. This cohort study included 56 ROS1+ patients (FISH or NGS confirmed); fusion partners were identified in 27 cases, including CD74 (n = 10), EZR (n = 7), and SDC4 (n = 7). Clinical data were available for 50 patients (median age 62; 51% female; 32% never-smokers). Forty patients received tyrosine kinase inhibitors (TKIs), mostly crizotinib (n = 38). Crizotinib showed a 55% objective response rate (ORR) and a median progression-free survival (mPFS) of 5.3 months. Brain metastases (HR 2.65, 95% CI 1.06–6.60, P = 0.037) and prior chemotherapy (HR 3.17, 95% CI 1.35–7.45, P = 0.008) had a higher risk of progression. Sixteen patients received subsequent lorlatinib, with an ORR of 28% and mPFS of 3.7 months. G2032R and L2026M resistance mutations were identified in four lorlatinib non-responders, and in vitro studies confirmed resistance to lorlatinib. Fusion partners did not affect crizotinib outcomes. Lorlatinib was ineffective against on-target resistance. Real-world data showed lower TKI efficacy than clinical trials, highlighting the role of clinical and molecular factors in treatment response.
DOCUMENT
BACKGROUND: T lymphocytes are orchestrators of adaptive immunity. Naïve T cells may differentiate into Th1, Th2, Th17 or iTreg phenotypes, depending on environmental co-stimulatory signals. To identify genes and pathways involved in differentiation of Jurkat T cells towards Th1 and Th2 subtypes we performed comprehensive transcriptome analyses of Jurkat T cells stimulated with various stimuli and pathway inhibitors. Results from these experiments were validated in a human experimental setting using whole blood and purified CD4+ Tcells.RESULTS: Calcium-dependent activation of T cells using CD3/CD28 and PMA/CD3 stimulation induced a Th1 expression profile reflected by increased expression of T-bet, RUNX3, IL-2, and IFNγ, whereas calcium-independent activation via PMA/CD28 induced a Th2 expression profile which included GATA3, RXRA, CCL1 and Itk. Knock down with siRNA and gene expression profiling in the presence of selective kinase inhibitors showed that proximal kinases Lck and PKCθ are crucial signaling hubs during T helper cell activation, revealing a clear role for Lck in Th1 development and for PKCθ in both Th1 and Th2 development. Medial signaling via MAPkinases appeared to be less important in these pathways, since specific inhibitors of these kinases displayed a minor effect on gene expression. Translation towards a primary, whole blood setting and purified human CD4+ T cells revealed that PMA/CD3 stimulation induced a more pronounced Th1 specific, Lck and PKCθ dependent IFNγ production, whereas PMA/CD28 induced Th2 specific IL-5 and IL-13 production, independent of Lck activation. PMA/CD3-mediated skewing towards a Th1 phenotype was also reflected in mRNA expression of the master transcription factor Tbet, whereas PMA/CD28-mediated stimulation enhanced GATA3 mRNA expression in primary human CD4+ Tcells.CONCLUSIONS: This study identifies stimulatory pathways and gene expression profiles for in vitro skewing of T helper cell activation. PMA/CD3 stimulation enhances a Th1-like response in an Lck and PKCθ dependent fashion, whereas PMA/CD28 stimulation results in a Th2-like phenotype independent of the proximal TCR-tyrosine kinase Lck. This approach offers a robust and fast translational in vitro system for skewed T helper cell responses in Jurkat T cells, primary human CD4+ Tcells and in a more complex matrix such as human whole blood.
DOCUMENT
Background: Adverse outcome pathway (AOP) networks are versatile tools in toxicology and risk assessment that capture and visualize mechanisms driving toxicity originating from various data sources. They share a common structure consisting of a set of molecular initiating events and key events, connected by key event relationships, leading to the actual adverse outcome. AOP networks are to be considered living documents that should be frequently updated by feeding in new data. Such iterative optimization exercises are typically done manually, which not only is a time-consuming effort, but also bears the risk of overlooking critical data. The present study introduces a novel approach for AOP network optimization of a previously published AOP network on chemical-induced cholestasis using artificial intelligence to facilitate automated data collection followed by subsequent quantitative confidence assessment of molecular initiating events, key events, and key event relationships. Methods: Artificial intelligence-assisted data collection was performed by means of the free web platform Sysrev. Confidence levels of the tailored Bradford-Hill criteria were quantified for the purpose of weight-of-evidence assessment of the optimized AOP network. Scores were calculated for biological plausibility, empirical evidence, and essentiality, and were integrated into a total key event relationship confidence value. The optimized AOP network was visualized using Cytoscape with the node size representing the incidence of the key event and the edge size indicating the total confidence in the key event relationship. Results: This resulted in the identification of 38 and 135 unique key events and key event relationships, respectively. Transporter changes was the key event with the highest incidence, and formed the most confident key event relationship with the adverse outcome, cholestasis. Other important key events present in the AOP network include: nuclear receptor changes, intracellular bile acid accumulation, bile acid synthesis changes, oxidative stress, inflammation and apoptosis. Conclusions: This process led to the creation of an extensively informative AOP network focused on chemical-induced cholestasis. This optimized AOP network may serve as a mechanistic compass for the development of a battery of in vitro assays to reliably predict chemical-induced cholestatic injury.
DOCUMENT

For the future circular economy, renewable carbon feedstocks manifest considerable promise for synthesizing sustainable and biodegradable polyhydroxyalkanoate (PHA). In this study, 16 wt% and 30 wt% PHA (cell dry weight) are respectively produced by thermophilic Caldimonas thermodepolymerans from beechwood xylan and wheat arabinoxylan as the sole carbon source. Moreover, an in silico study of the potential xylan-degrading proteins was conducted using proteome sequencing and CAZyme specialized bioinformatic tools. This study demonstrates the feasibility of utilizing complex polysaccharide substrates for PHA biosynthesis, thereby potentially eliminate additional processing steps and reducing overall production costs for sustainable plastic.
MULTIFILE